01/02/21
Vie et santé
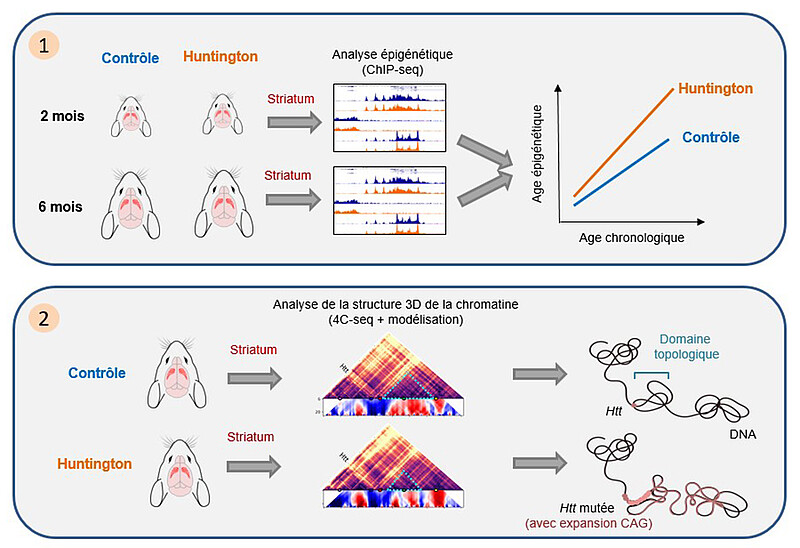
La maladie de Huntington est une maladie génétique rare. Entraînant la mort au terme d’une évolution de 10-15 ans, elle se caractérise par des symptômes moteurs (des mouvements involontaires) et comportementaux typiques, résultant d’une atteinte primaire des neurones du striatum, une région sous-corticale du cerveau. La dégénérescence des neurones du striatum chez les patients Huntington survient au terme d’une longue période de dysfonctionnement, portant atteinte à leur identité. On savait que l’expression des gènes de l’identité striatale et l’acétylation de la chromatine associée étaient réduites chez les patients Huntington et chez des souris modèles symptomatiques.
Cette nouvelle étude portée par le Laboratoire de neurosciences cognitives et adaptatives (LNCA) révèle que la perte sélective d’acétylation des histones au niveau des gènes de l’identité striatale se met en place très précocement, bien avant l’apparition des déficits moteurs chez la souris, et que ce processus a pour effet une accélération des mécanismes épigénétiques liés au vieillissement. La maladie de Huntington est causée par une mutation unique, une répétition anormale de codons CAG dans le gène Huntingtine (HTT). Les scientifiques montrent que des altérations épigénétiques ciblent plus spécifiquement le gène muté. Les résultats indiquent que la présence de la mutation chez la souris modifie précocement l’organisation tridimensionnelle de la chromatine et l’expression des gènes associés. Ces travaux améliorent ainsi la compréhension du processus pathogénique de la maladie de Huntington, en montrant que la mutation en cause affecte précocement la chromatine du striatum par deux mécanismes distincts.